

Anvisa aprova novo regulamento para registro de medicamentos biossimilares
A Anvisa aprovou, na reunião da Diretoria Colegiada desta segunda-feira (27/5), o novo regulamento para registro de medicamentos biossimilares.
O objetivo da nova norma é simplificar o processo de desenvolvimento desses produtos, a partir da flexibilização segura de requisitos. O regulamento irá possibilitar a dispensa de algumas etapas e estudos específicos, quanto tecnicamente viável, promovendo assim um ambiente regulatório transparente e previsível ao setor.
Os pontos e requisitos para a comprovação da comparabilidade entre os produtos foram exaustivamente discutidos durante o processo regulatório. Uma das novidades do novo regulamento é a possibilidade de utilização de medicamento de referência comparador adquirido em território internacional em situação de indisponibilidade e comprovados os requisitos técnicos necessários.
A norma hoje aprovada traduz o reconhecimento da Anvisa à importância do acesso dos pacientes aos produtos biológicos, já que a utilização dos biossimilares é uma estratégia de saúde pública importante para diminuir os custos dos medicamentos e aumentar a acessibilidade aos produtos biológicos e às novas tecnologias, com segurança e eficácia.
Processo regulatório
A nova regulamentação é resultado de estudos iniciados em 2022, com a publicação do Edital de Chamamento 15/2022, que colheu importantes informações e subsídios para o desenvolvimento de produtos biológicos pela via da comparabilidade.
Após o Edital, a Anvisa realizou um Diálogo Setorial com a sociedade civil, em 31 de julho do ano passado, que permitiu mais uma coleta de informações para a abertura do processo regulatório.
A norma foi construída de forma transparente, com grande participação da sociedade civil nas discussões desde o início do processo, por meio do Edital de Chamamento, Diálogo Setorial e Consulta Pública, etapas essenciais para o entendimento e a melhoria da proposta regulatória.
A Anvisa é uma das primeiras agências reguladoras a adequar o marco regulatório harmonizado internacionalmente com as principais agências reguladoras do mundo. A nova regra é resultado de um movimento mundial de adequação do marco regulatório para biossimilares, que teve sua importância ressaltada por todos os diretores da Agência para a ampliação do acesso a medicamentos biológicos.
Veja o voto da diretora supervisora Meiruze de Sousa Freitas.
Veja o voto do diretor relator Antonio Barra Torres.
Fonte: Site da ANVISA
Read MoreEsclarecimentos sobre a transparência das filas de análise na Anvisa
Aumento no volume de processos priorizados tem impactado as filas.
Recentemente, a área de Medicamentos da Anvisa recebeu alguns questionamentos sobre o ritmo das filas de análise de medicamentos genéricos e similares. Em resposta, a Agência reitera seu compromisso primordial de garantir a segurança, a eficácia e a qualidade dos produtos submetidos à sua análise.
Destaca-se o contínuo esforço para aprimorar os sistemas e procedimentos da Anvisa, visando mais eficiência e transparência em suas atividades. Como exemplo, temos a publicação recente de um painel detalhando as filas internas de cada unidade organizacional envolvida na avaliação de estudos necessários para o registro de medicamentos sintéticos e semissintéticos. Além disso, foi disponibilizado um relatório de gestão ao público, demonstrando a evolução das análises e dos pedidos de registros recebidos pela Agência.
Apesar disso, ainda é relevante contextualizar o aumento significativo no volume de processos priorizados, especialmente na categoria de genéricos, que tem impactado diretamente as análises de pedidos não priorizados de medicamentos genéricos e similares. Embora avanços notáveis tenham sido alcançados na regularização desses casos prioritários, o crescente volume de solicitações dessa natureza tem sobrecarregado os recursos disponíveis para a análise de outros processos. Confira abaixo dois gráficos que ajudam a visualizar a situação.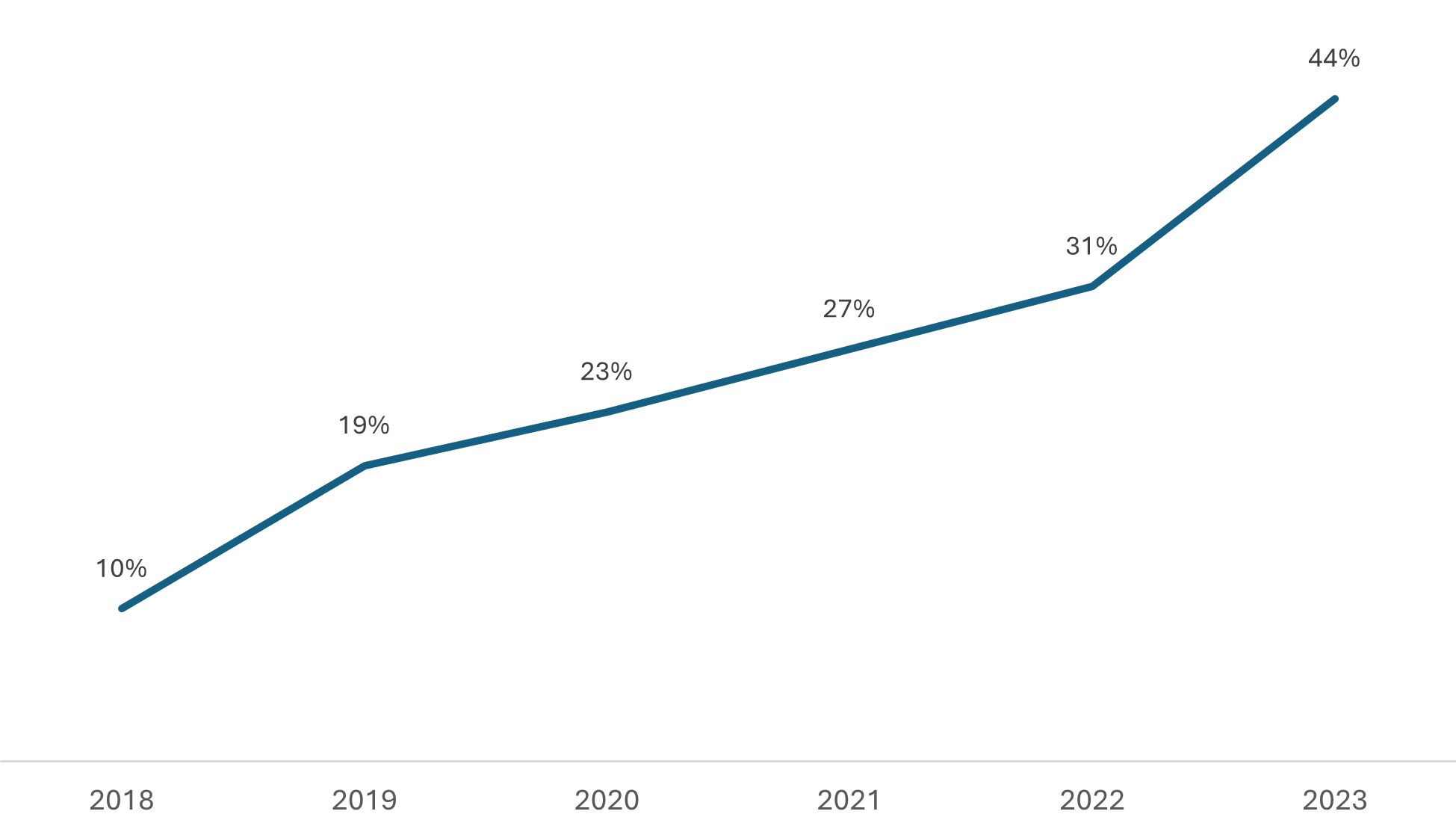 Ocupação da Capacidade de Análise da Gerência de Avaliação da Qualidade de Medicamentos (GQMED) com Processos Priorizados, considerando os recursos humanos atualmente disponíveis.
Ocupação da Capacidade de Análise da Gerência de Avaliação da Qualidade de Medicamentos (GQMED) com Processos Priorizados, considerando os recursos humanos atualmente disponíveis.
O Gráfico 1 mostra a ocupação da capacidade de análise da Gerência de Qualidade em Medicamentos (GQMED) com processos de genéricos priorizados ao longo do tempo.
Observa-se a variação na carga de trabalho e como isso tem afetado a capacidade de análise da GQMED. 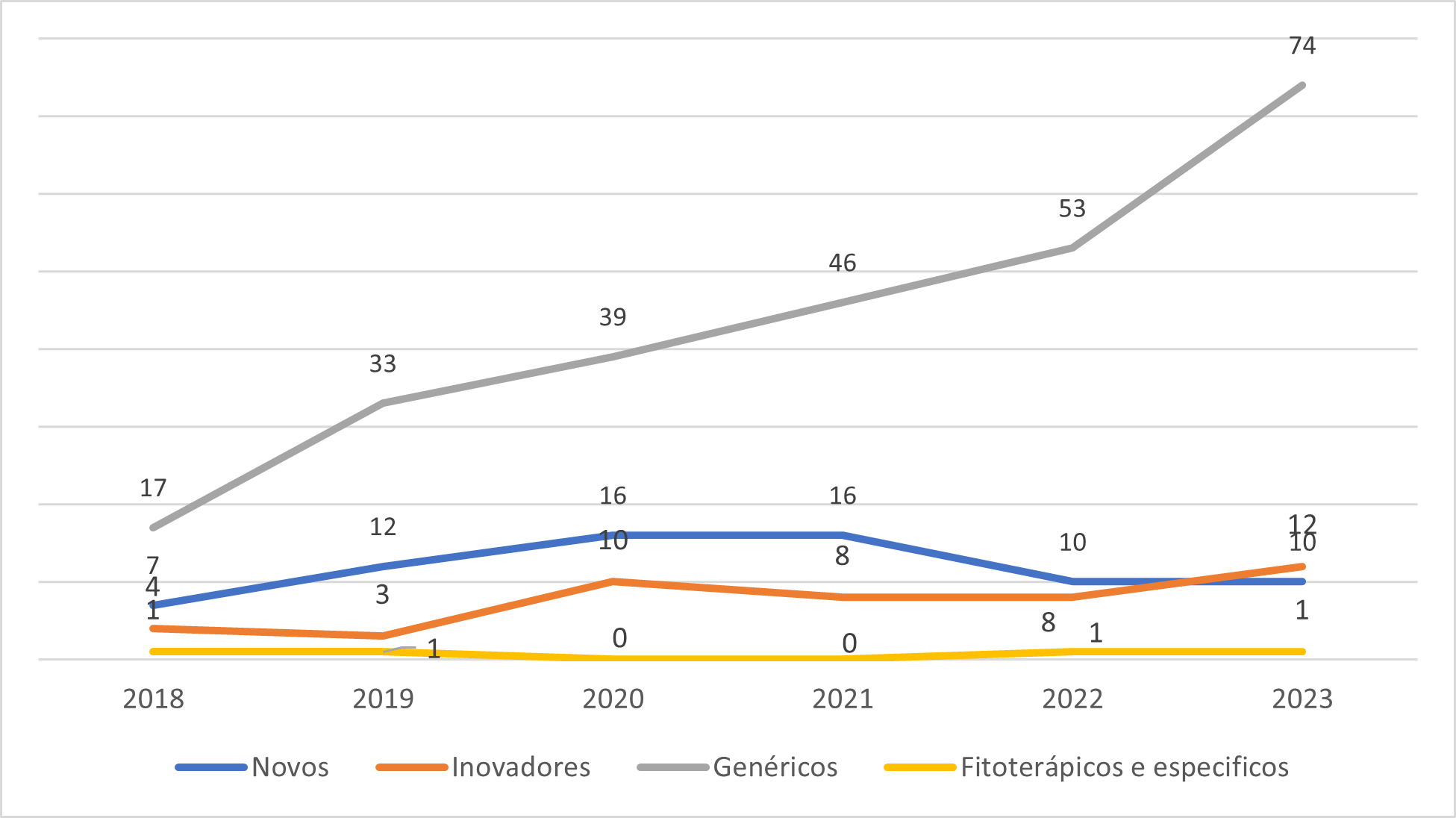 Evolução dos Pedidos de Registros de Medicamentos que tiveram análises priorizadas nos termos das Resoluções RDC 204 e 205, ambas de 2017.
Evolução dos Pedidos de Registros de Medicamentos que tiveram análises priorizadas nos termos das Resoluções RDC 204 e 205, ambas de 2017.
O Gráfico 2 ilustra a evolução dos pedidos de priorização de todos os registros de medicamentos sintéticos e semissintéticos. Destaca-se o aumento significativo na demanda por priorização, especificamente de medicamentos genéricos, o que tem impactado diretamente a dinâmica das análises realizadas pela Anvisa. Esse incremento acentuado também foi observado no ano de 2024, no qual o volume de processos de genéricos priorizados já representa 60% do total previsto para ser analisado no primeiro trimestre.
Portanto, se não houver aumento no número de técnicos responsáveis pelas análises e se for mantido o ritmo de crescimento, que em análise inicial indica um direcionamento da indústria para esse nicho de genéricos priorizados, é esperado que a velocidade de início das análises dos pedidos de registros de medicamentos genéricos e similares não priorizados continue diminuindo.
Por fim, a Anvisa reitera seu compromisso em atuar de forma diligente e responsável na análise dos processos, sempre visando o interesse público e a segurança dos cidadãos.
Esta notícia está clara para você? Clique aqui e responda nossa pesquisa em menos de 1 minuto. Categoria
Saúde e Vigilância Sanitária
Tags: medicamentos filas de análise genéricos similares registro
Fonte: Site da ANVISA
Read MoreAnvisa participa da 47ª reunião do ICH
A Anvisa irá participar da 47ª reunião do Conselho Internacional de Harmonização de Requisitos Técnicos para Produtos Farmacêuticos de Uso Humano (International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use – ICH). As atividades ocorrerão em Praga, na República Tcheca, entre os dias 28 de outubro e 1º de novembro de 2023, ocasião em que se reunirão a Assembleia, o Comitê Gestor e 14 Grupos de Trabalho.
Atualmente, a Agência participa de 27 Grupos de Trabalho e de três Grupos de Discussão do ICH, sendo representada por 52 especialistas no total. Para a reunião de Praga, a delegação brasileira terá 18 representantes, designados conforme os documentos de Governança do Conselho e a Portaria Anvisa 1520/2019, que trata do modelo de atuação regulatória para a incorporação de temas desenvolvidos pelo ICH. Veja a lista completa abaixo.
O ICH é uma iniciativa que reúne autoridades reguladores e a indústria farmacêutica para discutir aspectos científicos e técnicos do desenvolvimento e registro de produtos farmacêuticos. Além do Brasil, são membros as autoridades reguladoras da União Europeia, Estados Unidos, Japão, Canadá, Suíça, México, Egito, Singapura, República da Coreia, Reino Unido, China, Arábia Saudita, Taipé Chinesa e Turquia.
Em 2019, a Anvisa foi eleita para o Comitê Gestor do ICH, que tem como função propor à Assembleia a deliberação dos temas prioritários para harmonização e treinamento, a supervisão dos grupos de trabalho, bem como as decisões de governança do Conselho.
Paralelamente às reuniões do ICH, também se reunirá em Praga o Programa Internacional de Reguladores Farmacêuticos (International Pharmaceutical Regulators Programme – IPRP). O IPRP tem como objetivo favorecer o intercâmbio de informações e discussões de interesse comum entre os reguladores membros, promovendo a cooperação entre eles.
Delegação da Anvisa para a 47ª reunião do ICH
| Reunião | Representante |
| Reguladores, Assembleia, Comitê Gestor e IPRP | Nélio Cezar de Aquino |
| Reguladores, Assembleia, Comitê Gestor e IPRP | Bianca Zimon Giacomini Ribeiro |
| Reguladores, Assembleia, Comitê Gestor e IPRP | Varley Dias Sousa |
| Reguladores, Coordenadores, Assembleia, Comitê Gestor e IPRP | Ana Carolina Moreira Marino Araujo |
| Q1-Q5A | Renata Cristina Eto |
| Q5A (R2) | Silmara Cristiane da Silveira Andreoli |
| Q2(R2)/Q14 | João Tavares Neto |
| Q9(R1) | Nathalie Dias Kuwabara |
| E2D(R1) | Flavia Moreira Cruz |
| E6(R3) | Monica da Luz Carvalho Soares |
| E11A | Priscila Lemos Costa |
| E20 | Carolina Pingret Cintra |
| E21 | Christiane Santiago Maia |
| M1 Ptc | Juliana de Castro Zoratto |
| M4Q(R2) | Ellen Nogueira |
| M11 | Cláudio Nishizawa |
| M12 | Luiza Novaes Borges |
| M13 | Eduardo Agostinho Freitas Fernandes |
Categoria
Saúde e Vigilância Sanitária
Tags: reunião internacional ICH produtos farmacêuticos desenvolvimento registro harmonização regulatória cooperação internacional
Fonte: Site da ANVISA
Read More
INPI inicia recepção e processamento dos pedidos de registro para marca de posição
Os pedidos de registro para marca de posição já podem ser depositados no INPI. Com a criação de um formulário específico e as atualizações dos sistemas para recepção e processamento destes pedidos, o INPI encerra o trabalho de planejamento e implantação dos procedimentos necessários ao registro de marca de posição no Brasil.
As medidas adotadas permitirão que a Diretoria de Marcas, Desenhos Industriais e Indicações Geográficas dê andamento aos pedidos já depositados e que estavam aguardando as atualizações do sistema de processamento, conforme previsto no art. 95 da Portaria/INPI/PR nº 08/2022.
A informatização e o processamento das marcas de posição representam também o cumprimento da iniciativa estratégica número 7 do Plano de Ação INPI 2022.
Fonte: Site do INPI
Read MoreMedicamentos: novo fluxo para petições de aditamento
A Anvisa informa que, a partir do próximo dia 1º de novembro, será implantado um novo fluxo para petições de aditamento de processos de registro e pós-registro de medicamentos, insumos farmacêuticos ativos (IFAs) e de pesquisa clínica. A partir de então, essas petições deverão ser protocolizadas por meio do peticionamento eletrônico, via Sistema Solicita, utilizando os códigos de assunto listados ao final.
Resultado do aprimoramento do trabalho desenvolvido pela área de Medicamentos da Agência, o objetivo da medida é dar mais agilidade a essas demandas, bem como padronizar os procedimentos.
Com o envio exclusivamente eletrônico dessas petições, o expediente será imediatamente aditado ao processo, acelerando sua análise, principalmente nos casos em que a petição à qual se refere já estiver em análise. Ressalta-se que os aditamentos não serão considerados quando a análise da petição a que se refere já estiver finalizada (análise concluída).
É importante ressaltar ainda que estes assuntos podem ser vinculados a petições primárias ou secundárias e que a correta vinculação é feita pela empresa durante o peticionamento. Dessa forma, vincular o aditamento ao expediente correto é de suma importância para a localização da complementação enviada.
De acordo com a RDC 204/2005, os aditamentos devem ser utilizados para toda e qualquer complementação ao processo, não exigida formalmente, que se limita ao aprimoramento do conhecimento do objeto do processo, não resultando em manifestação diversa da peticionada. Dessa forma, destaca-se que um aditamento nunca deverá ser utilizado como petição de pós-registro; caso contrário, isso poderá prejudicar a empresa, por exemplo, em uma análise fiscal do produto.
Confira abaixo quais são os códigos de assuntos que deverão ser peticionados nesse novo fluxo:
| 10118 | CENTRO DE EQUIVALÊNCIA FARMACÊUTICA – Aditamento |
| 11516 | CERTIFICAÇÃO DE BOAS PRÁTICAS DE BIODISPONIBILIDADE/BIOEQUIVALÊNCIA – MEDICAMENTOS – Aditamento |
| 1620 | DINAMIZADO – Aditamento |
| 1363 | ENSAIOS CLÍNICOS – Aditamento |
| 1741 | ESPECÍFICO – Aditamento |
| 1952 | GENÉRICO – Aditamento |
| 1719 | MEDICAMENTO FITOTERÁPICO – Aditamento |
| 1432 | MEDICAMENTO NOVO – Aditamento |
| 1913 | PRODUTO BIOLÓGICO – Aditamento |
| 11554 | PRODUTO DE CANNABIS – Aditamento |
| 10635 | PRODUTO TRADICIONAL FITOTERÁPICO – Aditamento |
| 10366 | RADIOFÁRMACO – Aditamento |
| 10322 | Registro de IFA – Aditamento (Petições de Registro, Pós-Registro e Renovação) |
| 1416 | SIMILAR – Aditamento |
Fonte: Site da ANVISA
Read MoreMedicamento matriz em adequação não pode ter fármaco clone vinculado
A Anvisa informa às empresas do setor de fármacos que não serão aceitas petições clones relacionadas a medicamentos matriz que estejam em fase de adequação relacionada à comprovação de qualidade, segurança ou eficácia do produto. A regra está descrita no artigo 6º da Resolução da Diretoria Colegiada (RDC) 31/2014.
Sendo assim, serão indeferidas (negadas) as petições de registro de medicamentos clones vinculados a produtos matriz cujo registro foi concedido mediante a apresentação de termo de compromisso.
Petição clone
Um medicamento clone é aquele que possui a mesma linha de produção, mesmo fabricante, mesmos relatórios técnico e clínico e a mesma composição de outro medicamento já registrado junto à Anvisa, denominado matriz. Neste caso, os clones são diferentes apenas em aspectos como nome e rotulagem, por exemplo.
Fonte: Site da ANVISA
Read MoreParticipe da elaboração das perguntas e respostas sobre o Guia 24/2019
A Anvisa abriu nesta quarta-feira (11/8) o prazo para o envio de contribuições do setor regulado para a elaboração das perguntas e respostas sobre o Guia 24/2019, que trata da organização do Documento Técnico Comum (Common Technical Document – CTD) para registro e pós-registro de medicamentos.
O objetivo é incentivar as empresas da indústria farmacêutica a particiar deste processo e a utilizar o formato CTD. Todas as contribuições recebidas serão avaliadas e poderão subsidiar a revisão do Guia e a consequente publicação de uma nova versão do documento.
Para participar, basta acessar e preencher o formulário eletrônico criado para o registro das sugestões. O prazo final para enviar contribuições é o dia 8 de setembro deste ano. Participe!
Melhores práticas
O Guia 24/2019 expressa o entendimento sobre as melhores práticas com relação a procedimentos, rotinas e métodos considerados adequados ao cumprimento de requisitos técnicos ou administrativos exigidos pelos marcos legislativo e regulatório da Agência, referentes ao CTD para registro e pós-registro de medicamentos no Brasil.
Trata-se de um instrumento regulatório não normativo, ou seja, seu caráter é recomendatório. A inobservância do seu conteúdo não caracteriza infração sanitária nem é motivo para indeferimento de petições, uma vez atendidos os requisitos exigidos pela legislação.
Quer saber as notícias da Anvisa em primeira mão? Siga-nos no Twitter @anvisa_oficial, Facebook@AnvisaOficial, Instagram @anvisaoficial e YouTube @anvisaoficial
Categoria
Saúde e Vigilância Sanitária
Tags: medicamentos guia 24/2019 perguntas e respostas documento técnico comum ctd registro pós-registro
Medicamentos sintéticos: novos códigos para petição
A Anvisa informa que os novos códigos de assuntos para petição secundária de registro e pós-registro de medicamentos sintéticos já estão disponíveis. Portanto, os códigos antigos serão excluídos do sistema de peticionamento eletrônico.
Também já foram disponibilizados os documentos do seminário virtual realizado em 5/7 sobre o novo fluxo de publicação para medicamentos sintéticos, inclusive o texto que reúne as principais perguntas e respostas sobre o tema. Os materiais estão reunidos neste link. É importante observar que as perguntas e respostas serão atualizadas periodicamente. O recebimento de contribuições pode ser realizado por meio de formulário específico.
Confira abaixo as alterações:
Registro:
|
Códigos antigos |
Códigos novos |
|
11627 GENÉRICO – Formulário de informações relativas à documentação de registro (FIDR) ou Parecer de Análise Técnica da Empresa (Pate) |
12107 GENÉRICO – Formulário de informações relativas à documentação de registro (FIDR) – Análise de Qualidade Registro |
|
11626 MEDICAMENTO INOVADOR – Formulário de informações relativas à documentação de registro (FIDR) ou Parecer de Análise Técnica da Empresa (Pate) |
12108 MEDICAMENTO INOVADOR – Formulário de informações relativas à documentação de registro (FIDR) – Análise de Qualidade Registro |
|
11625 MEDICAMENTO NOVO –Formulário de informações relativas à documentação de registro (FIDR) ou Parecer de Análise Técnica da Empresa (Pate) |
12109 MEDICAMENTO NOVO – Formulário de informações relativas à documentação de registro (FIDR) – Análise de Qualidade Registro |
|
11628 SIMILAR – Formulário de informações relativas à documentação de registro (FIDR) ou Parecer de Análise Técnica da Empresa (Pate) |
12110 SIMILAR – Formulário de informações relativas à documentação de registro (FIDR) – Análise de Qualidade Registro |
Pós-registro:
|
Códigos antigos |
Códigos novos |
|
11627 GENÉRICO – Formulário de informações relativas à documentação de registro (FIDR) ou Parecer de Análise Técnica da Empresa (Pate) |
12111 GENÉRICO – Parecer de Análise Técnica da Empresa (Pate) – Análise de qualidade Pós-Registro |
|
11626 MEDICAMENTO INOVADOR – Formulário de informações relativas à documentação de registro (FIDR) ou Parecer de Análise Técnica da Empresa (Pate) |
12112 MEDICAMENTO INOVADOR – Parecer de Análise Técnica da Empresa (Pate) – Análise de qualidade Pós-Registro |
|
11625 MEDICAMENTO NOVO – Formulário de informações relativas à documentação de registro (FIDR) ou Parecer de Análise Técnica da Empresa (Pate) |
12113 MEDICAMENTO NOVO – Parecer de Análise Técnica da Empresa (Pate) – Análise de qualidade Pós-Registro |
|
11628 SIMILAR – Formulário de informações relativas à documentação de registro (FIDR) ou Parecer de Análise Técnica da Empresa (Pate) |
12114 SIMILAR – Parecer de Análise Técnica da Empresa (Pate) – Análise de qualidade Pós-Registro |
Quer saber as notícias da Anvisa em primeira mão? Siga-nos no Twitter @anvisa_oficial, Facebook@AnvisaOficial, Instagram @anvisaoficial e YouTube @anvisaoficial
Categoria
Saúde e Vigilância Sanitária
Tags: medicamentos sintéticos registro pós-registro peticionamento eletrônico novos códigos de assuntos
Fonte: Site da ANVISA
Read MoreCovid-19: norma deve agilizar registro de produtos
A Anvisa definiu normas extraordinárias para avaliação de pedidos de registro de medicamentos e produtos biológicos para prevenção e tratamento do novo coronavírus (Covid-19). As regras também estabelecem procedimentos extraordinários para alteração pós-registro, quando a empresa faz mudança no registro original do medicamento ou produto biológico. Além disso, há mudança extraordinária para o registro de produtos para a realização de diagnóstico laboratorial (in vitro) do vírus.
O objetivo da medida é ampliar opções de prevenção e tratamento, bem como evitar o desabastecimento de produtos. Visa também aumentar as alternativas de detecção da doença. A ideia não é avaliar e aprovar produtos de forma automática, pois o rigor sanitária sempre deve existir. Mas, sim, dar mais rapidez ao processo de análise de pedidos.
As normas estão na Resolução de Diretoria Colegiada (RDC) 348/2020, publicada do Diário Oficial da União (D.O.U) desta quarta-feira (18/3), data em que as regras passam a vigorar, com prazo de validade de seis meses.
Informações gerais
De acordo com a resolução, o registro de medicamentos e produtos biológicos só será concedido quando ficar configurada a indicação terapêutica específica para prevenção ou tratamento da doença causada pelo Covid-19 ou diagnóstico in vitro (laboratorial) para SARS-CoV-2.
Já no caso dos pedidos de mudanças pós-registro, o medicamento deverá ser considerado essencial para manutenção da vida ou utilizado em caso de grave risco à saúde. Entre outros critérios, também está a ameaça de desabastecimento de produtos comprovadamente ligada ao novo coronavírus.
Leia na íntegra a Resolução de Diretoria Colegiada (RDC) 348/2020.
Quer saber as notícias da Anvisa em primeira mão? Siga-nos no Twitter @anvisa_oficial, Facebook@AnvisaOficial, Instagram @anvisaoficial e YouTube @anvisaoficial
Read MoreMudanças no processo de registro reduzem fila
O ano de 2017 começou com um grande desafio para a Anvisa: cumprir os prazos da Lei 13.411/2016, publicada em 28 de dezembro do ano passado. Há um ano, a nova lei definiu prazos de 120 dias para o registro de medicamentos prioritários e 365 para processos ordinários, sendo que naquele momento o prazo de similares e genéricos ultrapassava facilmente esses tempos.
Se 2017 foi desafio, 2018 será a hora da recompensa, com a perspectiva de zerar a fila e adequar todos os processos aos novos prazos da lei. A expectativa é de que, em alguns meses, todas as petições de medicamentos genéricos e similares estejam dentro do prazo.
A nova regra provocou uma reflexão sobre como as atividades na área de medicamentos eram realizadas e o impacto disso para a sociedade e a economia do país. Apesar dos avanços promovidos em anos anteriores no processo de avaliação de medicamentos, a Lei 13.411/2016 exigiu da equipe da Gerência-Geral de Medicamentos (GGMED) uma transformação mais radical e impactante.
Tratar o passivo da Anvisa em 365 dias foi uma tarefa ousada. Os processos existentes foram separados em quatro grupos. Da divisão realizada em julho de 2017 em grupos de análise de genéricos e similares, por similaridades, atualmente temos 13 processos no grupo I, 60 processos no grupo II, oito processos no grupo III e 363 processos no grupo IV, totalizando 444 processos na fila para análise.
Essa ação faz parte do P1 do Planejamento Estratégico da Anvisa e deve ser expandida para outras áreas de registro de produtos em 2018.
Todos os colaboradores da Anvisa, especialmente a equipe da GGMED, continuam empenhados para que a estratégia traçada seja cumprida. Em 2018 haverá ainda mais trabalho.
#2018SemFila
Fonte: Site da ANVISA
Read More